


Gene therapy slows Huntington’s disease — a breakthrough rewriting hope for neurodegenerative medicine.
In a landmark development, researchers report that a single administration of a novel gene therapy, AMT-130, produced a 75 % slowing of disease progression over three years in patients with Huntington’s disease. This achievement marks the first time a therapy has demonstrably altered the course of this genetic brain disorder — and signals a turning point for the field of neurological gene therapy.
The Landmark Trial and Its Key Findings
In September 2025, uniQure and collaborators disclosed top-line results from a Phase I/II clinical trial of AMT-130, a gene therapy aimed at Huntington’s disease. Over three years, patients receiving the high dose of AMT-130 (administered via neurosurgical infusion) experienced about 75 % slower disease progression, as measured via standardized clinical scales comparing treated vs external controls.
The primary endpoint was met: treated individuals declined much more slowly across domains of motor control, cognition, and functional abilities. The trial used the cUHDRS (composite Unified Huntington’s Disease Rating Scale) metric, showing that the treated group lost on average ~0.38 points, versus ~1.52 points in matched historical controls.
Notably, the therapy also showed biomarkers consistent with neuroprotection: treated patients had lower levels of neurofilament light chain (a marker of neuronal damage) than controls, suggesting that fewer neurons were undergoing active degeneration.
Because Huntington’s is an inherited, progressive neurodegenerative disorder long considered untreatable, this result is hailed as historic. As one researcher put it: “This result changes everything.”
However, it’s important to emphasize: AMT-130 is not a cure. Rather, it is the first to convincingly slow — not stop or reverse — the disease. The trial involved a relatively small number of participants, and full peer review and longer follow-up are needed.
How AMT-130 Works: A Closer Look
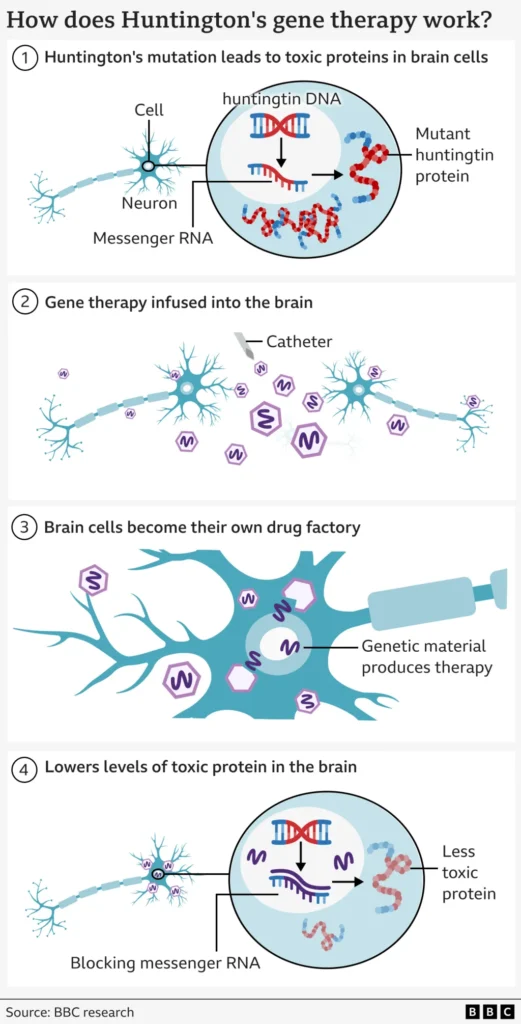
Targeting the Root: Reducing Toxic Huntingtin Expression
Huntington’s disease (HD) is caused by a mutation in the HTT gene, specifically an expanded number of trinucleotide repeats (CAG) in the gene’s coding region. This mutation leads to the production of a mutant huntingtin protein that aggregates and causes progressive neuronal damage, particularly in regions like the striatum and cortex.
AMT-130 uses a viral vector (AAV5) as a delivery vehicle — essentially a benign “Trojan horse” virus engineered to carry a microRNA (miRNA) cassette. Once inside neurons, the miRNA targets the messenger RNA (mRNA) transcripts encoding huntingtin (both mutant and some wild-type), thereby reducing production of the toxic protein.
Because this is delivered directly to brain tissues (via neurosurgical infusion to specific regions), the therapy bypasses the blood–brain barrier and ensures sufficient concentration in target neurons. The effect is expected to last long-term (ideally, one treatment for many years).
Risks, Delivery Challenges, and Safety Considerations
Delivering therapy directly to the brain is inherently high risk. The infusion is a lengthy neurosurgical procedure (12–20 hours), requiring microcatheter placement into bilateral brain regions. Risks include hemorrhage, infection, inflammation, and off-target effects.
In the trial, some serious adverse events occurred in the high-dose arm, prompting safety reviews and adjustments. This caution is understandable given the irreversible nature of gene therapies once delivered.
Moreover, because the miRNA approach targets both mutant and some non-mutant huntingtin mRNA, there remains a theoretical concern about reducing too much normal huntingtin, which might have biological roles. Researchers must carefully balance efficacy with safety.
Finally, long-term durability remains unanswered. We don’t yet know whether the effect lasts a decade or more, or whether compensatory biological pathways will reduce efficacy over time.
Comparison with Other Experimental Approaches
AMT-130 isn’t the only line of attack against Huntington’s — but it is the first to produce a disease-modifying effect in humans.
Small Molecules / Splicing Modulators
- Votoplam (PTC518) — an oral splicing modulator designed to reduce HTT mRNA levels by forcing inclusion of a “pseudoexon” that triggers mRNA decay. Early trials have shown modest reductions in huntingtin protein.
- Branaplam (LMI070 / NVS-SM1) — initially developed for spinal muscular atrophy, later repurposed for HD. However, its clinical trial in HD was discontinued due to toxicity signals.
- Pridopidine — though not a direct huntingtin-lowering agent, it modulates the sigma-1 receptor pathway and shows neuroprotective potential. It’s being explored for symptomatic benefits in HD.
These small-molecule/drug approaches tend to require repeated dosing and may have lower brain penetrance, but they are non-invasive and easier to scale if efficacy and safety prove acceptable.
Alternative Gene Therapies
Other gene therapy strategies seek to modulate the neuronal environment or metabolism rather than directly lowering huntingtin. For instance, AB-1001 is an experimental gene therapy aimed at raising CYP46A1 enzyme levels (thus modifying cholesterol metabolism in the brain) as a supplementary strategy in HD.
CRISPR / Genome Editing
The broader field is watching with interest whether CRISPR or base editing methods might one day directly correct the HTT gene mutation. The Economist’s article alludes to “clever genetic techniques” that may underpin future therapies.
However, genome editing in human brains raises enormous technical, ethical, and safety challenges (off-target edits, immune response, and delivery). For now, silencing/knockdown approaches like AMT-130 are more mature.
Regulatory and Commercial Outlook
The success of AMT-130 is already creating momentum on the regulatory front. UniQure is expected to file for U.S. approval in early 2026, following further data submission and review.
Previously, AMT-130 received RMAT (Regenerative Medicine Advanced Therapy) designation from the FDA, which accelerates regulatory interactions and review pathways for potentially transformative therapies.
From a financial perspective, the announcement triggered a dramatic investor response: uniQure’s stock surged over 200 %, and its partner ClearPoint (which supplies the infusion catheter) also saw gains.
But commercial challenges remain: cost, scalability, reimbursement, and equitable access are major obstacles — especially in low- and middle-income settings. The high price of neurosurgical delivery and long-term monitoring may limit uptake unless payers and governments step in.
Broader Implications and Future Directions
A Template for Neurodegenerative Gene Therapies
The success of AMT-130 signals that genetic brain diseases once deemed untreatable might now be within reach. The result may catalyze more investment into gene therapy for Alzheimer’s, Parkinson’s, ALS, and others, with more advanced vectors, delivery systems, and editing tools.
Already, Nature has noted that “brain editing is closer to reality,” citing other experimental gene-editing tools that show promise in animal models. Nature
Improved Early Diagnosis and Genetic Screening
As effective therapies emerge, patients and at-risk individuals may be more willing to get genetic testing early. This may shift Huntington’s from an inevitably fatal diagnosis to a condition managed with preventive intervention. The Guardian
Ethical, Social, and Access Questions
With radical new therapies come harder questions: who gets access first? How do we handle long-term follow-up and unknown side effects? Will this widen therapeutic inequality globally?
Especially for rare diseases like HD, patient advocacy groups, insurers, and public health agencies will need to collaborate to ensure fair distribution and post-market safety monitoring.
Next Steps in Clinical Research
- Expanded cohorts & longer follow-up: The existing trial is small; larger, multi-center studies with longer durations will help confirm durability and safety.
- Refined vector and delivery strategies: Optimizing viral tropism, reducing off-target effects, improving ease of infusion, and minimizing risk.
- Combining strategies: Future regimens might combine gene therapy with small-molecule drugs or neuroprotective agents to optimize outcomes.
- Exploring other neurological diseases: The techniques refined in Huntington’s may become the blueprint for tackling other inherited brain disorders.
Challenges, Limitations & Caveats
- Small sample size and external controls — The trial’s reliance on historical matched controls rather than randomized placebo groups introduces potential biases.
- Safety and adverse events — Neurosurgical risks, immune reactions, and unintended gene suppression remain unresolved.
- Durability unknown — It’s unclear how long suppression of huntingtin will persist; compensatory biology could dampen the effect over many years.
- Cost and access — Without heavy support, many patients may not access therapy in low-resource settings.
- Biological complexity — The brain is a complex, interconnected organ. Slowing neuron loss doesn’t necessarily reverse prior damage, and functional recovery may remain limited for some patients.
Despite these caveats, the milestone is real: for the first time, Huntington’s disease progression has been meaningfully slowed in humans.
Conclusion
This gene therapy breakthrough marks a turning point in the fight against Huntington’s disease — and perhaps in the broader domain of neurologic genetic disorders. AMT-130’s ability to slow degeneration gives hope beyond symptom management, toward disease modification.
Still, much work lies ahead. Regulatory review, long-term safety, access equity, and further trials must all succeed for this hope to fully materialize. But the scientific community now knows it’s no longer a dream — slowing a genetic brain disease is possible.
If we get the next steps right — inclusive access, responsible oversight, and alliance between researchers, regulators, and patient communities — AMT-130 could become a template for how we treat previously hopeless neurological conditions.
Subscribe to trusted news sites like USnewsSphere.com for continuous updates.


